转录组分析概述
本文最后更新于 2025年2月7日 下午
概述
转录组(transcriptome)广义上指某一生理条件下,细胞内所有转录产物的集合,包括信使RNA、核糖体RNA、转运RNA及非编码RNA;狭义上指所有mRNA的集合。转录组测序分析可以分为有参转录组分析和无参转录组分析。有参无参的意思是,有/无参考基因组。
转录组上游分析
转录组上游分析一般有3种模式:
- 有参转录组上游分析
- 有参考基因组无注释文件上游分析或寻找新的转录本
- 无参考基因组
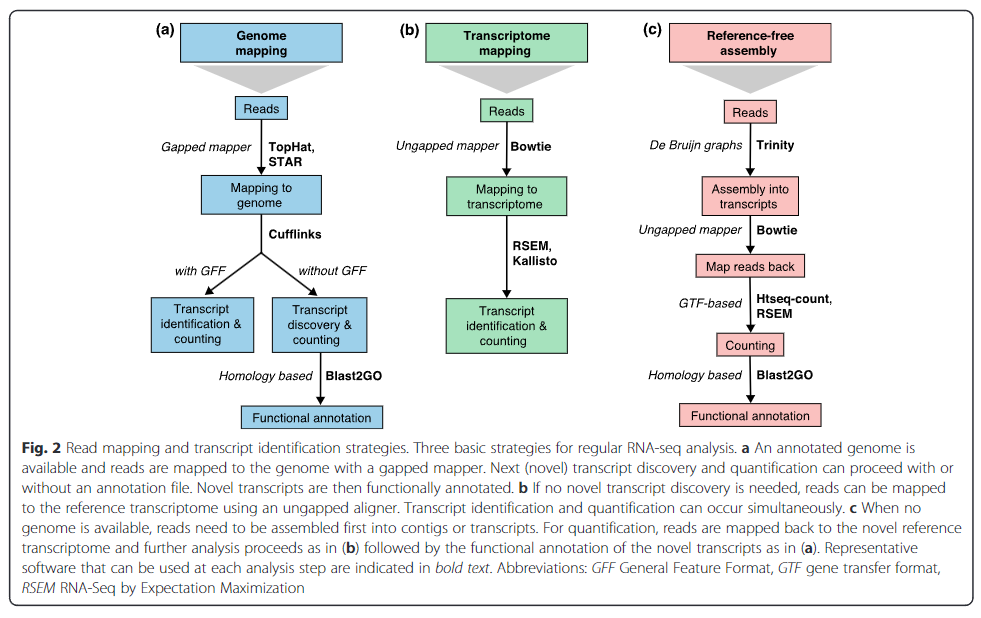
DOI: 10.1186/s13059-016-0881-8
测序数据及质量控制
测序数据主要由fastq格式提供,包含测序片段的序列信息和相应的测序质量打分,每条read由四行描述信息组成:
1 | |
- 第一行以
@开头,后面包含测序标识符和描述文字 - 第二行为测序片段的序列信息
- 第三行以
+开头,后面为测序标识符 - 第四行为测序片段的质量值。
测序数据过滤
使用过滤软件保留高质量的序列数据,去除接头序列,参考过滤标准:
- 去除带接头(adapter)的 reads;
- 当任一测序read中N含量超过该read碱基数的10%时,去除此paired reads;
- 当任一测序read中含有的低质量(Q<=20)碱基数超过该条read碱基数的50%时,去除此paired reads。
测序错误分布
当前RNA-seq测序技术,测序错误率分布存在以下两个特征:
- 测序错误率随着测序序列(Sequenced Reads)长度的增加而升高。这是由测序过程中化学试剂的消耗导致的;
- 前6个碱基具有较高的测序错误率,此长度恰好为RNA-seq建库过程中反转录所需的随机引物长度。前6个碱基测序错误率较高是因为随机引物和RNA模版的不完全结合
GC含量分布
GC含量分布检查用于检测有无 AT、GC 分离现象。由于序列的随机打断和双链互补原则,理论上测序读段在每个位置的 GC 及 AT 含量应分别相等,且在整个测序过程基本稳定不变。但由于反转录使用的是 6 bp 随机引物,因此前几位碱基在核苷酸组成上会有一定偏好性,产生正常波动,随后则趋于稳定。
转录组下游分析
进行差异表达基因的筛选,也可将转录本比对到参考序列后进行基因结构方面的分析。
转录组生物信息分析流程如下图所示:
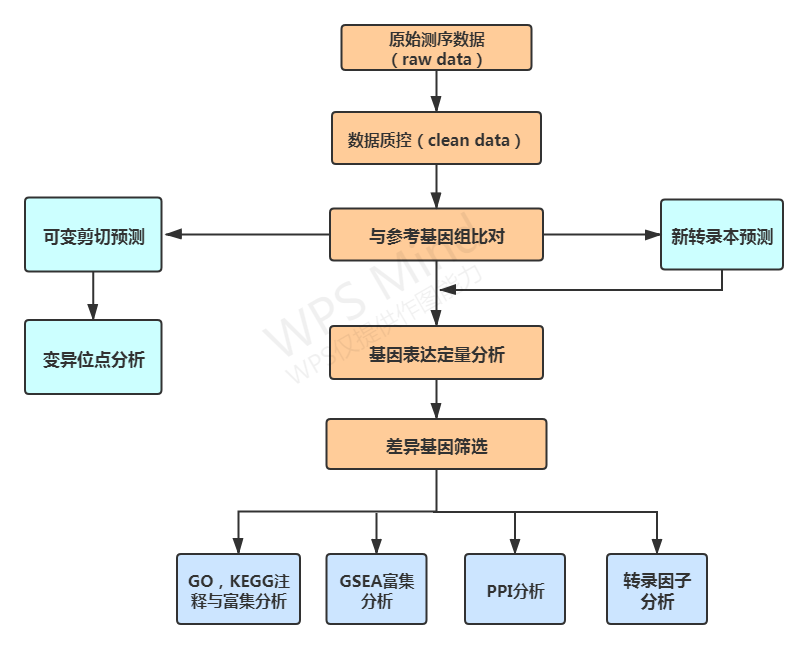
新转录本预测
根据比对上基因组的 reads 的位置信息,使用StringTie将 reads 组装成转录本。 StringTie 应用网络流算法和可选的从头组装(denovo)来拼接转录本。拼接得到的转录本再使用 GffCompare 与基因组的注释信息比较,从中发现新转录本或新基因。
可变剪切鉴定
可变剪接(Alternative Splicing, AS),是大多数真核生物细胞中普遍的一种基因表达方式。真核细胞的基因序列包含内含子(intron)与外显子(exon),在基因转录成mRNA前体后内含子会被RNA剪接体移除,而外显子则保留于成熟mRNA中。一条未经剪接的RNA,可以具有多种外显子剪接形式,因此使得一个基因在不同时间、不同环境中可以翻译出不同的蛋白质,进而增加其生理状况下系统的复杂性或适应性。转录组数据的可变剪接分析使用rMATS实现
SNP/InDel检测
单核苷酸多态性(Single Nucleotide Polymorphism,SNP)指基因组上由单个核苷酸变异形成的遗传标记。SNP 标记具有多态性丰富、分布广、遗传稳定性强等特点,而且可以实现高通量、自动化检测,具有低成本、高效的优点,因而成为研究生物表型与基因型之间关系的重要桥梁。插入/缺失(Insertion-Deletion,InDel)是指在近缘种或同一物种不同个体之间基因组同一位点的序列发生了不同大小核苷酸片段的插入或缺失,即一个序列上某一位点相比同源的另一个序列插入或缺失了一个或多个碱基。InDel 标记因其稳定性好、多态性高、分型系统简单,已广泛应用于动植物群体遗传分析、分子辅助育种等领域。一般而言,SNP 是指变异频率大于 1 % 的单核苷酸变异,InDel 的长度大部分在 50 bp 以内。
由于部分 mRNA 会经历 RNA 编辑(RNA editing),即转录后的 RNA 在编码区发生碱基的插入、删除或替换,产生多态性的基因表达产物。从比对结果来看,SNP 和单碱基替换的 RNA 编辑结果相同。因此通过转录组测序数据检测到的 SNP 和 InDel 难免含有 RNA 编辑的产物。SNP 和 InDel的检测使用GATK实现,注释使用ANNOVAR实现
基因功能注释
从基因组中提取基因的序列,使用 diamond将新基因与 KEGG、GO、NR、Swiss-Prot、TrEMBL、KOG 数据库序列比对得到注释结果,比对条件为Evalue 1e-5。植物转录因子预测使用iTAK软件,iTAK 整合了两个数据库,PlnTFDB 和 PlantTFDB。动物转录因子鉴定使用动物转录因子数据库animalTFDB
基因组注释(三):基因功能注释 | 生信技工 (yanzhongsino.github.io)
基因表达定量
根据比对结果和基因在参考基因组上的位置信息,统计每个基因的reads数量。一个转录本的片段数目与测序数据(或Mapped Data)量、转录本长度、转录本表达水平都有关,为了让片段数目能真实地反映转录本表达水平,需要对样品中的Mapped Reads的数目和转录本长度进行归一化。
差异表达分析
GO功能显著性富集分析,KEGG信号通路富集分析,WCGNA,聚类分析,PCA
**DESeq2、edgeR 和 limma ** 三大包可以说是做转录组差异分析的金标准,大多数转录组的文章都是用这三个 R 包进行差异分析的。
首先,做差异分析需要的数据有:表达矩阵和分组信息。
DESeq2、edgeR 和 limma 包都建议使用 Counts,也就是原始计数数据作为输入进行分析,最好不要使用 FPKM、TPM 等归一化后的数据
参考:DEG-workshop