基因组结构注释
本文最后更新于 2025年2月7日 下午
基因组注释分为结构注释和功能注释。
结构注释能够帮助我们获取基因组上详细的基因分布和结构信息,是功能注释和进化分析的重要基础。基因结构预测包括预测基因组中的基因位点、开放性阅读框架(ORF)、翻译起始位点和终止位点、内含子和外显子区域、启动子、可变剪切位点以及蛋白质编码序列等等。
功能注释可以帮助我们进一步获得基因的功能信息,包括预测基因中的模序和结构域、蛋白质的功能和所在的生物学通路等
基因结构注释流程:
注意:在开始基因组结构注释前,最好将所有碱基全部改成大写形式,因为小写碱基有些软件是识别成soft-masked的形式,影响注释预测,edta的TIR预测和geta软件都有可能报错。
1 | |
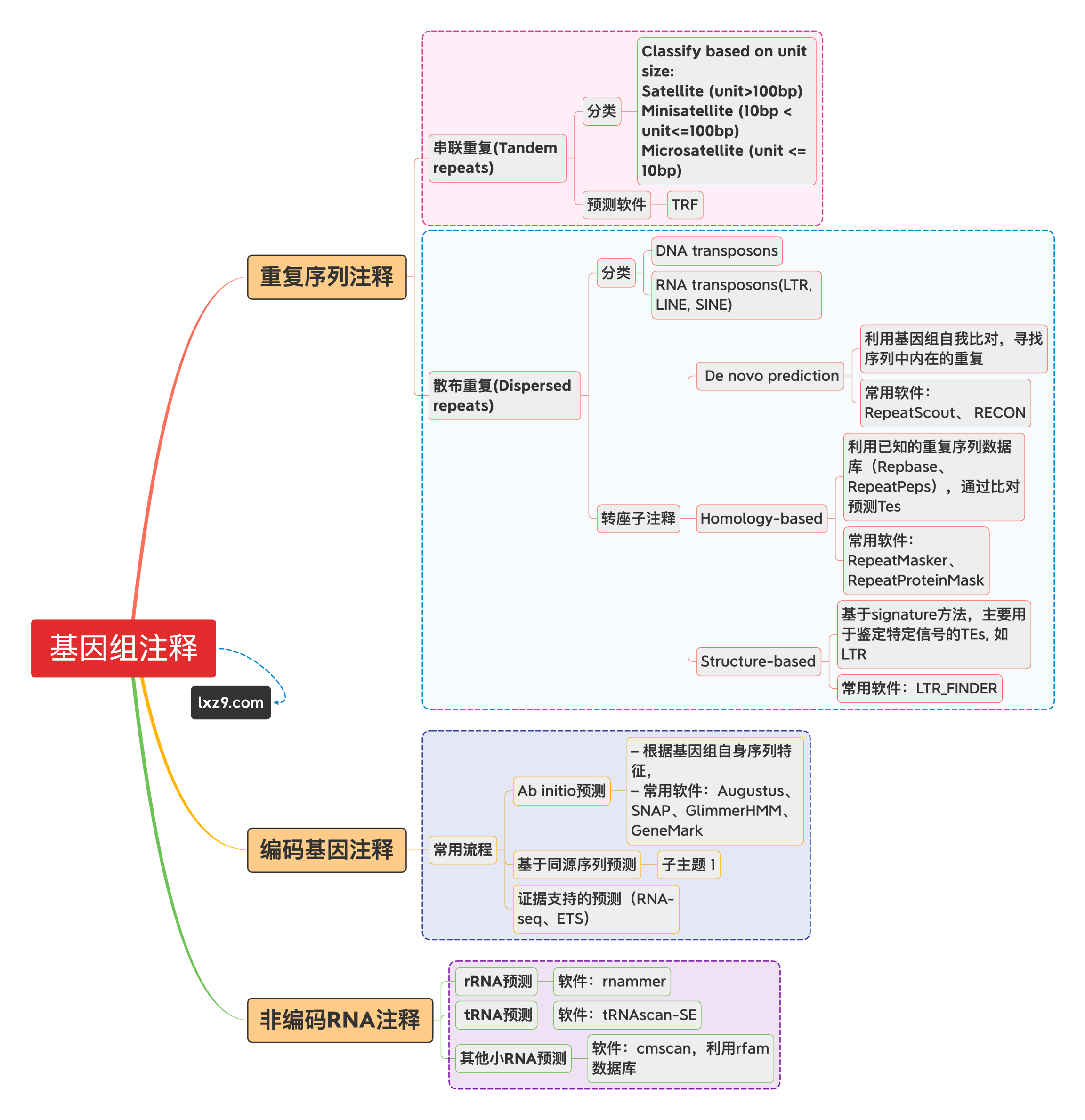
重复序列注释
❗❗❗ 重复序列的注释非常重要,如果跳过这一步,会对基因组结构注释的准确性造成很大影响
重复序列在基因组中占有非常高的比例,根据分布将重复序列分为串联重复(Tandem repeats)和散布重复(Dispersed repeats)
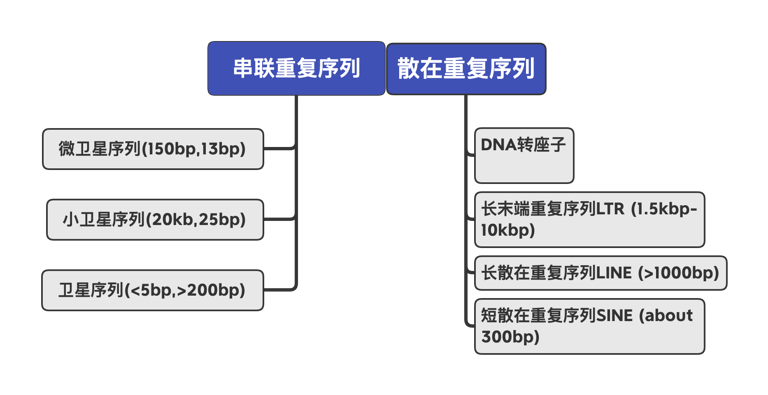
对重复序列的注释一般是做基因组注释的第一步,常用的基因组重复序列注释软件有RepeatMasker, RepeatModeler, EDTA
大部分情况下ReapeatMasker的数据库中没有待注释物种的近缘种,此时最好用RepeatModeler等软件进行自我训练,用denovo预测来构建重复序列数据库。即使数据库中已有近缘种,有时候Repbase注释重复区的效果不是很好,也推荐进行自我训练。
这里首先使用RepeatModeler对基因组重复序列进行自我训练,从头预测重复序列,构建数据库。随后使用RepeatMasker对基因组重复序列进行标注
RepeatMasker, RepeatModeler的安装
这两个软件都不推荐从源码安装,安装依赖多的容易让人怀疑人生。
这里提供两种安装方式
conda安装
1 | |
这里推荐指定两个conda chanel, bioconda和conda-forge,提高安装成功率。有很多依赖包在conda-forge里才能找到。这里同样也推荐新建conda环境,用于这两个软件的安装。
mamba是conda的C++语言版本替代,可以大幅提高conda的响应时间。mamba-org/mamba: The Fast Cross-Platform Package Manager (github.com)
docker安装
这里使用了由Dfam团队维护的docker镜像进行安装,也可以在github中克隆项目,自己构建镜像。Dfam-consortium/TETools: Dfam Transposable Element Tools Docker container. (github.com)
1 | |
- 这里使用了
-v选项来将宿主目录挂载到容器内,如果需要对RepeatModeler的Libraries进行更改,就可以先将相关目录挂载到容器内,再对容器内的/opt/RepeatMasker/Libraries目录进行修改。 - Dfam团队的官方镜像很大,解压过后更是达到了90GB以上,可以考虑自己从源码构建镜像。
RepeatModeler的使用
建库
1 | |
自我训练
1 | |
- 总共跑6轮,前4轮有几分钟到几十分钟的
-threads超负荷运行,后两轮每轮会有长达十几个小时的-threads超负荷运行 - 自我训练过程需要耗费数天
- 如果基因组太大,可能会出现
sample.translation文件读取错误的问题,这个时候可以将基因组文件分成几个小文件(大概4GB左右,已测试),对每个小文件的重复序列进行从头预测,然后将结果进行合并。Could not open *.translation file for reading! · Issue #248 · Dfam-consortium/RepeatModeler (github.com)
结果文件:
- sample-families.fa (用于
RepeatMasker指定lib) - sample-families.stk(用于上传到Dfam数据库)
- sample-rmod.log(程序运行日志文件)
使用seqkit对.fa文件中重复的id重命名
1 | |
RepeatMasker的使用
1 | |
-species选择RepeatMasker数据库中已有的近缘物种-pa线程数量,用RMBlast引擎会用到4核,即设置-pa 12会用到48线程。-lib指定RepeatModeler自我训练的库sample-families.fa-poly在file.poly中保存多态的polymorphic的简单重复序列,即微卫星重复序列。-html输出xhtml格式文件-gff输出gff格式文件-dir指定输出文件的目录,默认时当下目录
⚠️⚠️⚠️注意:RepeatMasker的运行非常占用内存资源,如果内存不够,可以考虑将基因组按照scaffold或chromosome拆分文件,运行结束后再将结果进行合并。
结果:
- sample.fa.tbl:结果报告,统计了基因组长度,GC含量,重复序列长度及各类重复序列占比等。
- sample.fa.out:将基因组中预测得到的重复序列和参考序列相比的碱基替换频率、插入/删除率,以及重复序列的位置、结构、类型等信息展示出。
- sample.fa.out.gff:sample.fa.out的gff格式。
- sample.fa.out.html:sample.fa.out的网页形式。
- sample.fa.polyout:通过-poly参数,额外将sample.fa.out中的微卫星注释提取出来。微卫星注释是一种Simple_repeat序列,可以通过*.polyout将*.out的微卫星注释删除。有研究者认为微卫星不能算作严格的重复序列类型,还有研究者甚至认为Simple_repeat全都不能算。
- sample.fa.cat.ga:记录了基因组序列和数据库中参考重复序列的比对详情,其中“i”和“v”分别代表了碱基转换(transitions)和颠换(transversions),“-”表示该位点存在碱基插入/删除。
- sample.fa.masked:将重复序列屏蔽成N碱基的masked基因组。
sample.fa.out每一列含义:
- 第一列:比对分值,SW score
- 第二列:替代率 perc div.
- 第三列:碱基缺失百分率
- 第四列:在重复序列中碱基缺失百分率
- 第五列:query sequence
- 第六列:查询序列起始位置
- 第七列:查询序列终止位置
- 第八列:查询区域中超出比对区域碱基的数- 目,也就是没有比对上的碱基数
- 第九列:+/-©
- 第十列:比上的重复序列名称,类型命名
- 第十一列:比上重复序列的分类,和- repeatmolder 中*.classed 是一样的
- 第十二列:比上的在数据库中的起始位置
- 第十三列:比上的在数据库中的终止位置
- 第十四列:在第十列上超出比对区域碱基的数目,也就是没有比对上的碱基数
- 第十五列:比对区域的ID,随机给的
编码基因注释
这里采用BRAKER对基因组编码基因进行注释
BRAKER提供了4种运行模式:
- BRAKER with RNA-Seq data
- BRAKER with protein data
- BRAKER with RNA-Seq and protein data
- BRAKER with short and long read RNA-Seq and protein data
- 使用高质量的基因组
- 使用简单的scaffold (contig, chromosome) 名称
- 屏蔽重复序列
- 调整运行参数以适应不同的基因结构
- 在使用注释之前评估注释结果
建议docker安装 teambraker/braker3 - Docker Image | Docker Hub
1 | |
安装后运行容器shell
1 | |
在容器shell中进行编码基因注释
1 | |
- 这里为第三种运行模式
--bam参数后必须为.bam文件,如有多个文件,需要用逗号隔开
结果:
- braker.aa 从注释中提取的蛋白质序列
- braker.codingseq 从注释中提取的CDS序列
- braker.gtf BRAKER预测的基因组结构文件
- hintsfile.gff 来自RNA-Seq/蛋白数据的外部证据信息